| [ Team LiB ] |
|
6.2 AlignmentsThe alignments and alignment statistics reported by BLAST differ slightly from program to program. The rest of this chapter describes the details of BLASTP, BLASTN, BLASTX, TBLASTN, and TBLASTX alignments and shows how to recognize alignment groups. 6.2.1 BLASTPBLASTP alignments are the simplest to understand. Figure 6-2 shows the anatomy of a typical BLASTP alignment. Figure 6-2. A BLASTP alignment Here are the parts you should pay attention to:
The database sequence may be several lines long if the BLAST database is a nonredundant database with concatenated definition lines. For more on this topic, see Chapter 11. The WU-BLAST format differs slightly from the NCBI format: gaps aren't reported on the statistics line, and the P-value (displayed as P or Sum P) is always reported in addition to the Expect. 6.2.2 BLASTNDNA is a double-stranded molecule, and genes may occur on either strand. This fact makes BLASTN alignments a little more difficult to interpret than BLASTP alignments. When a query sequence is searched against a database, both strands of the query are examined. The plus strand is the sequence in the FASTA file. The minus strand is the reverse complement of this sequence. If the similarity between the query and subject sequences is on the same strand, both sequences are labeled as being on the plus strand and the coordinates increase from left to right (Figure 6-3a). Since BLAST just aligns letters and has no model of genes or other features, it is impossible to determine on which strand a gene lies from a BLASTN alignment. Even if an alignment is labeled as "Plus/Plus," the encoded gene may be on the minus strand. When the minus strand of the query sequence is similar to a database sequence, the alignment is reported with either the subject or query sequence in reversed coordinates. In NCBI-BLAST, the database sequences are flipped (Figure 6-3b), but in WU-BLAST, the query coordinates are flipped (Figure 6-3c). Figure 6-3. BLASTN alignments: (a) NCBI-BLAST, same strand; (b) NCBI-BLAST, different strand; (c) WU-BLAST, different strand Table 6-1 shows how strand is displayed in the five standard BLAST programs.
Here are a few minor notes:
6.2.3 BLASTXAlignments from BLASTX are complicated by both strand and reading frame. The query sequence is translated in three frames on both the plus and minus strands. Chapter 2 discusses the reading frame in more detail. With three nucleotides per codon, the coordinates of the query sequence increase by threes (Figure 6-4a). On the plus strand, the reading frame is computed relative to the start of the plus strand; reading frame 1 starts at position 1 and reading frame 2 starts at position 2. On the minus strand, the reading frame is calculated relative to the reverse complement of the plus strand; the last letter of the FASTA file starts frame -1 and the second-to-last letter starts frame -2. Minus strand matches invert the query coordinates (Figure 6-4b). Figure 6-4. BLASTX alignments (ovals indicate that nucleotide coordinates increase by threes (a) and are reversed for minus strand matches (b))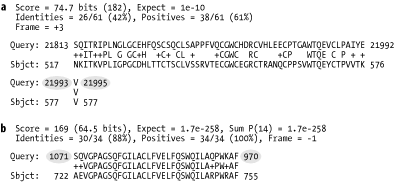 6.2.4 TBLASTNTBLASTN alignments are very similar to BLASTX alignments, except that the database and query are exchanged. Therefore, the database sequence increases in threes, and the database sequence has flipped coordinates on the minus strand. 6.2.5 TBLASTXTBLASTX has more complicated alignments because both the query and the database have strand and frame. Figure 6-5 shows examples of all strand combinations. One of the most confusing aspects of TBLASTX alignments is that a number of different frames may represent the same region from both the query and subject. A TBLASTX alignment between two genomic sequences often highlights shared coding sequences. However, the correct frame of the encoded proteins can't be determined from a TBLASTX report. Chapter 8 and Chapter 9 discuss techniques that make TBLASTX more discriminate. Figure 6-5. TBLASTX alignments (coordinates increase by threes and may have any combination of frames) 6.2.6 Alignment GroupsAlignment groups are one of the most confusing aspects of the BLAST report. Chapter 4 and Chapter 5 discuss how and why alignments are sometimes grouped to increase their statistical significance. However, the standard BLAST format doesn't make this structure easy to see. Figure 6-6 shows the scores reported for various alignments in a single database hit. The groups can be inferred from the Expect values. If several alignments have the same E-value, it is more difficult to determine which alignments belong to which groups. Figure 6-6. Alignment groups (groups can be inferred from Expect values) By default, WU-BLAST alignment groups are just as difficult to recognize as NCBI-BLAST groups. WU-BLAST has a very useful command-line option called topcomboN that organizes and limits the number of groups. Chapter 8 discusses topcomboN in more detail. Figure 6-7 shows how groups are organized by strand and then by Sum P-value for a single database hit. Groups are labeled and need not be inferred. Notice that some groups contain only one alignment. Figure 6-7. WU-BLAST alignment groups with topcomboN=9 |
| [ Team LiB ] |
|