| [ Team LiB ] |
|
2.3 Genomes and GenesIn general, the genomic structure of prokaryotes is very different from that of eukaryotes (Figure 2-5). Genomes are organized into chromosomes. Prokaryotes often have a single circular chromosome, and eukaryotes usually have multiple linear chromosomes. People are sometimes surprised to find that genome size and chromosome number aren't reflected in organismal complexity. For example, the single-celled Amoeba dubia has a genome that is about 200 times larger than the human genome. Although dogs and cats have very similar genome sizes, dogs have twice as many chromosomes. One rule to keep in mind when thinking about genomic organization is that genomes of viruses and prokaryotic organisms generally contain little noncoding sequence, whereas the genomes of more complex organisms usually contain a much higher percentage of noncoding sequence. Figure 2-5. Prokaryote and eukaryote cells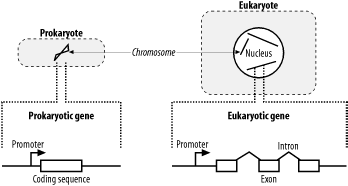 2.3.1 Prokaryotic GenesProkaryotic genes are relatively simple. They contain a promoter that determines when the gene is transcribed and a coding region that contains the DNA sequence for a protein. It is relatively easy to find genes in prokaryotic genomes. Since stop codons are expected about every 21 triplets (there are three stop codons out of 64 total triplet combinations), long open reading frames (ORFs) should be very rare, at least from an unbiased random model. On average, proteins are 300 amino acids long, so finding an ORF that is 900 nucleotides long is really unexpected and a pretty clear signal that the ORF codes for a real protein. Of course, some genes encode small proteins, and finding these is a bit more difficult. 2.3.2 Eukaryotic GenesEukaryotic gene structure is more complicated than prokaryotic gene structure. Unlike prokaryotic genes, eukaryotic genes are often broken into pieces that are assembled before they are translated. Like prokaryotes, eukaryotes also have promoters to regulate when genes are turned on, but they are often much larger and may exist a great distance from the start of translation. In addition, many genes respond to additional sequences called enhancers and suppressors that aren't necessarily upstream of a gene and may be many kilobases away. In eukaryotes, mRNAs are processed before they are translated (Figure 2-6). Two kinds of processing are common: splicing and poly-adenylation. Splicing brings together the coding sequences and throws out the intervening stuff. The sequences that end up in the mature mRNA are called exons, and the intervening stuff is called introns. The part of the mRNA that codes for protein is called the coding sequence (CDS), and the parts at either end are called untranslated regions (UTRs). The other common post-transcriptional modification is poly-adenylation. In this process, 50 or more adenine nucleotides are added to the end of the mRNA, which is called a poly-A tail. Figure 2-6. Eukaryotic mRNA processing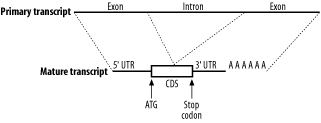 2.3.3 TranscriptsTo many people, the most interesting parts of a genome are its genes. However, genes may account for a small fraction of a genome. In the human genome, for example, only 1 to 2 percent of the sequence codes for proteins. So why not just sequence the proteins? This procedure turns out to be much more difficult than sequencing nucleotides, but you can sequence the transcripts that code for proteins. Using some clever molecular biology techniques, it's possible to separate mRNAs from the rest of the cellular material and in this way specifically select for protein-coding genes. However, the mRNAs aren't sequenced directly. First they are copied into complementary DNA (cDNA) by an enzyme called reverse transcriptase. This enzyme converts mRNA into DNA, flouting the first rule, which is the Central Dogma of Molecular Biology. A collection of cDNAs is called a cDNA library, and it is common to have cDNA libraries from many kinds of tissues. The mRNAs present in the liver may be very different from those in the brain (the tissues have very different properties due to different collections of proteins). If you're interested in certain cancers, for example, you might develop and sequence cDNA libraries derived from specific types of tumors. In the world of sequencing, it is therefore common to find cDNA sequencing projects in addition to, or instead of, genome sequencing projects. The downside to cDNA sequencing is that many interesting sequences aren't transcribed, and those that are transcribed may be difficult to capture if they aren't abundant. In your quest for jargon compliance, note that sequencing reads from cDNA sequences are often called expressed sequence tags (ESTs). You will probably come across this term frequently in your BLAST searches. 2.3.4 RepeatsRepeats are one of the most mysterious features of genomes. All genomes sequenced to date contain some form of repeat, but the big eukaryotic genomes are richest. About half the human genome is easily recognized as repetitive. Understanding repeats is critical to BLAST users because if they aren't dealt with properly, they can tie up your computer for days, dominate your report, invalidate your statistics, and obscure your findings. The words "repeat" and "repetitive sequence" are used very loosely in genomics, and this causes a lot of confusion for novices. Broadly speaking, repeats can be classified as simple and complex. Simple repeats generally consist of low-complexity sequences (see Chapter 4); examples include runs of a single nucleotide such as An, Tn, Gn, and Cn; dinucleotide repeats such as [CA]n; tri-nucleotide repeats in the form of [CAG]n; and so on. The strange thing about these sequences is that they occur much more frequently in genomes than you'd expect by chance. Simple sequence repeats occur just about everywhere in the genome, even in the protein coding exons of genes, but they are especially common in heterochromatic, pericentromeric, and telomericregions of eukaryotic chromosomes that play structural roles and don't contain many genes. The term complex repeat usually describes any genomic repeat that doesn't consist of low complexity/low entropy sequence. Noncoding RNAs, such as rRNAs and tRNAs, comprise one commonly encountered class of complex repeat, but because they have known important functions, they are often not lumped together with the rest of the repeats. The term complex repeat can also denote some form of mobile genetic element or selfish DNA (a phrase coined by Francis Crick). These entities are a bit like the fleas and ticks of the genome: they copy and spread themselves within and between genomes and are generally believed to do little for the host genome. Selfish DNAs are usually further classified into three subcategories: transposons, retroviruses, and retrotransposons. If you see these names in a BLAST report, you may need to use a repeat filter. 2.3.5 PseudogenesOne of the most confounding problems in similarity searches is the presence of pseudogenes. As the name suggests, pseudogenes are "fake genes"; that is, they look like they could encode a protein, but they aren't functional. Pseudogenes come from a variety of sources. A mutation that introduces a stop codon into a gene creates a pseudogene, but more commonly, pseudogenes are created from some kind of duplication event. Sometimes, through various mechanisms, regions of a chromosome may become duplicated. The extra copies of genes are generally free of selective pressures and may become pseudogenes as they accumulate mutations. Duplication may also result from repetitive elements that include neighboring DNA as they copy themselves into new locations. In eukaryotes, a very common form of pseudogene is the retro-pseudogene, in which the mRNA from a gene is reverse-transcribed into DNA and inserted back into the genome. Because retro-pseudogenes come from mRNA, they contain the hallmarks of transcripts, notably an absence of introns and the presence of a poly-A tail. They are therefore easy to detect if you know what to look for. Most retro-pseudogenes come from highly transcribed genes such as the protein components of the ribosome. |
| [ Team LiB ] |
|